
Growing resistance to antibiotics and other antimicrobial treatments is a serious global healthcare challenge. A new study in Antibioticsdemonstrates a method for tracking the spread of genes for antimicrobial resistance among bacterial populations over time. The new computational technique relies on the rapidly increasing availability of bacterial genetic sequences in public databases such as GenBank.
“Our idea is that this could be used as a monitoring system,” explains Ivan Erill, professor of biological sciences at UMBC and the study’s senior author. “It’s great for studies trying to find insight into what’s happening in bacterial genomes.”
The code Erill and colleagues Miquel Sánchez-Osuna and Jordi Barbé at the Universitat Autònoma de Barcelona developed can analyze the sequences of all known bacterial plasmids (little circular pieces of DNA that can exchange genes between bacteria) in about an hour. The results reveal which resistance genes are spreading most and the genes’ likely origin.
A computational analysis like this is much faster and less expensive than complex systems involving coordination among clinicians around the world. This means it could be carried out more frequently to help doctors and researchers stay updated on shifting resistance threats.
“There’s going to be more and more data that you can mine this way,” Erill says, noting that the amount of genetic sequence data available is doubling approximately every two years. He adds, “I love it because it’s simple. It’s fast, and you can deploy it in a flash.”
 Ivan Erill (Marlayna Demond ’11/UMBC)
Ivan Erill (Marlayna Demond ’11/UMBC)
Genetic detective work
So how does this new technique work? Microbial DNA, like all DNA, is made up of four bases: A, T, G, and C. A pairs with T, and G pairs with C. However, the ratio of the bases varies considerably across microbial species. Some bacteria are split 50-50 between AT and GC pairs, while other bacterial genomes may contain anywhere from 30 to 70 percent GC pairs. In a previous study, Erill and colleagues leveraged this variability to investigate the emergence of resistance against sulfonamides, an early class of antimicrobials.
As resistance genes hop from species to species via plasmids, they largely retain the GC ratio of their original source. So, if there is a mismatch between the GC ratio of the resistance gene and the rest of a bacterium’s genome, that means the resistance gene has come from elsewhere. The simplicity of this technique means it is not only faster than clinical methods at tracking the movement of resistance genes, but also faster than other computational methods.
If a resistance gene has been in a species long enough, its genetic sequence may eventually begin to approach the GC content of its new host, but that could take millions of years. “For what we’re looking at, which is gene movement in the last 60 to 100 years,” Erill says, “it’s basically a snapshot.”
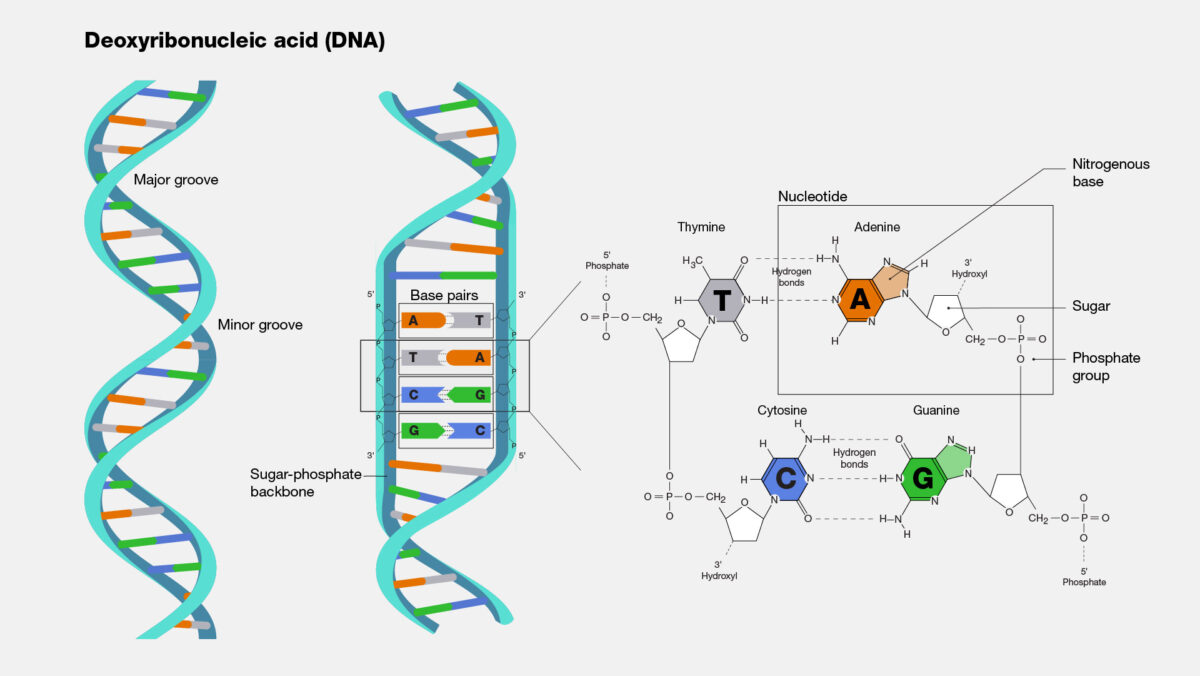 DNA is made up of adenine-thymine (AT) bonds and guanine-cytosine (GC) bonds. The frequency of each type of bond differs substantially across bacterial species. (Image by the National Human Genome Research Institute)
DNA is made up of adenine-thymine (AT) bonds and guanine-cytosine (GC) bonds. The frequency of each type of bond differs substantially across bacterial species. (Image by the National Human Genome Research Institute)
Specialists spread faster
Using the new monitoring technique, the study authors confirmed that resistance genes are most likely to spread if they are on conjugative plasmids, a type of plasmid that can easily transfer between bacterial cells. Researchers generally already understood this, but confirming it with the new method helped verify the technique’s efficacy.
The new study also found that resistance genes effectively targeting very specific antibiotics spread the most. These genes generally require so many mutations to evolve that they are unlikely to have arisen naturally in any given bacterium since humans started using antibiotics. But if they are present anywhere in the bacterial population when the corresponding antibiotic is introduced, they will spread quickly.
“As soon as there is selective pressure from that antibiotic, there is selective pressure to move this thing around, because it is a bacterium’s silver bullet against that antibiotic,” Erill says. In contrast, generic resistance that requires only a few mutations to existing genes is less likely to spread rapidly, Erill explains. “There isn’t a lot of selective pressure to pass it along, because by the time it comes, the bacterium has likely already discovered it,” he says.
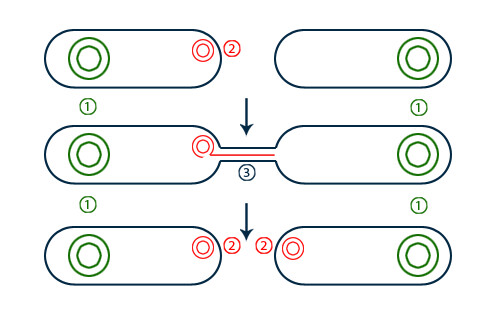 Bacterial cells (black outlines) contain plasmids (red) in addition to their main genome (green). A conjugative plasmid can transfer genetic material between cells as shown here, which can spread antibiotic resistance genes among species. (Image by Zappys Technology Solutions, used under CC-BY)
Bacterial cells (black outlines) contain plasmids (red) in addition to their main genome (green). A conjugative plasmid can transfer genetic material between cells as shown here, which can spread antibiotic resistance genes among species. (Image by Zappys Technology Solutions, used under CC-BY)
Hospitals aren’t likely the culprit
The new study also found that genes for resistance to antibiotics used in livestock or prescribed outside of hospitals were likely to spread through the global bacterial population. Resistance to antibiotics used in more limited settings hardly spread at all. “That tells you that if you use things cautiously, then there is not so much selective pressure,” Erill says.
Perhaps most important for antibiotic policy moving forward, Erill’s team found that most of the resistance genes came from a single source and then spread, rather than evolving independently multiple times. “Resistance is in the environment,” Erill says, explaining that it needs a vehicle to get into the mainstream.
If antibiotics were only used in hospitals, rather than in livestock and other environments, resistance would be much less common, Erill argues. This is because resistance from hospital use alone “would presume that you have naturally resistant bacteria living in the hospital already, ready to pass on their genes,” he suggests. While there are certainly infectious microbes present in hospitals, “most of the microbial diversity is in the soil and the water,” Erill says. If antibiotics never reach the cells that happen to be resistant, that resistance won’t spread.
 A tractor sprays an apple orchard. Antibiotics are regularly used on “top fruit” crops like apples, oranges, and pears, which can contribute to antibiotic resistance when the spray runs off into waterways and soil. (Photo by Barbara Eckstein, used under CC BY-NC-ND)
A tractor sprays an apple orchard. Antibiotics are regularly used on “top fruit” crops like apples, oranges, and pears, which can contribute to antibiotic resistance when the spray runs off into waterways and soil. (Photo by Barbara Eckstein, used under CC BY-NC-ND)
While the new study is “a methods paper more than a results paper,” Erill says, “we believe it’s an important contribution.” It puts forward a process for continually monitoring shifts in bacterial genomes over time, which could influence future antibiotic development research or treatment regimens. Perhaps it could even encourage limits on antibiotic use in agriculture and other settings where the drugs can end up in the environment.
Best of all, other research teams can use the new method to pursue answers to their own questions, Erill explains. “You can use it with a very fine comb to poke at whatever you are interested in.”





